Metastable atomic-ordered configurations for Al1/2Ga1/2N predicted by Monte-Carlo method based on first-principles calculations
-
Authors :
Jessiel Siaron Gueriba, Hiroshi Mizuseki, Marilou Cadatal-Raduban, Nobuhiko Sarukura, Yoshiyuki Kawazoe, Yosuke Nagasawa, Akira Hirano and Hiroshi Amano
-
Journal :
Journal of Physics: Condensed Matter
-
Vol :
36
-
Page :
13
-
Year :
2023
Abstract
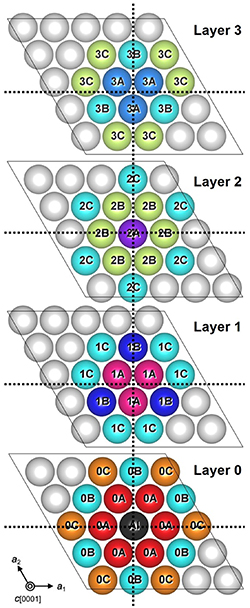
Metastability of Aln/12Ga1−n/12N (n = 2–10: integer) with the 1–2 monolayer (ML) in-plane
configuration towards the c [0001] direction has been demonstrated recently. To theoretically
explain the existence of these metastable structures, relatively large calculation cells are needed.
However, previous calculations were limited to the use of small calculation cell sizes to estimate
the local potential depth (∆σ) of ordered Al1/2Ga1/2N models. In this work, we were able to
evaluate large calculation cells based on the interaction energies between proximate Al atoms
(δEAl–Al) in AlGaN alloys. To do this, δEAl–Al values were estimated by first-principles
calculations (FPCs) using a (5a1 × 5a2 × 5c) cell. Next, a survey of the possible ordered
configurations using various large calculation cell models was performed using the estimated
δEAl–Al values and the Monte-Carlo method. Then, various ∆σ values were estimated by FPCs
and compared with the configurations previously reported by other research groups. We found
that the ordered configuration obtained from the (4a1 × 2a2 × 1c) calculation cell (C42) has the
lowest ∆σ of −9.3 meV/cation and exhibited an in-plane configuration at the c(0001) plane having (–Al–Al–Ga–Ga–) and (–Al–Ga–) sequence arrangements observed along the m
{
1¯100}
planes. Hence, we found consistencies between the morphology obtained from experiment and
the shape of the primitive cell based on our numerical calculations.