An assessment of density functionals for predicting CO 2 adsorption in diamine-functionalized metal-organic frameworks
-
Authors :
Jung-Hoon Lee,* Per Hyldgaard, Jeffrey B. Neaton*
-
Journal :
J. Chem. Phys
-
Vol :
156
-
Page :
154113
-
Year :
2022
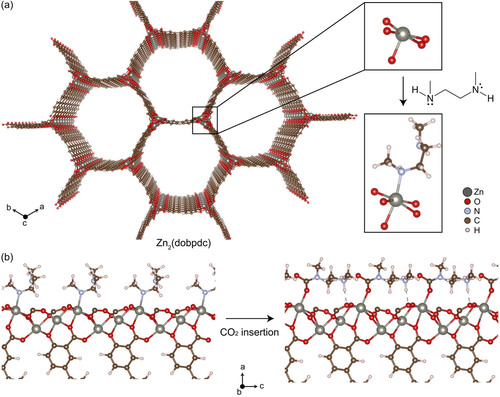
Abstract
Diamine-functionalized M2(dobpdc) (M = Mg, Mn, Fe, Co, Zn) metal-organic frameworks (MOFs) are among a growing class of crystalline solids currently being intensively investigated for carbon capture as they exhibit a novel cooperative and selective CO2 adsorption mechanism and a step-shaped isotherm. To understand their CO2 adsorption behavior, ab initio calculations with near-chemical accuracy (∼6 kJ/mol, an average experimental error) are required. Here, we present density functional theory (DFT) calculations of CO2 adsorption in m-2-m-Zn2(dobpdc) (m-2-m = N,N'-dimethylethyle-nediamine and dobpdc4- = 4,4'-dioxidobiphenyl-3,3'-dicarboxylate) with different exchange-correlation functionals, including semilocal functionals [Perdew-Burke-Ernzerhof (PBE) and two revised PBE functionals], semiempirical pairwise corrections (D3 and Tkatchenko-Scheffler), nonlocal van der Waals (vdW) correlation functionals-vdW-optB88 (or vdW-DF-optB88), vdW-DF1, vdW-DF2, vdW-DF2-B86R (or rev-vdW-DF2), vdW-DF-cx (and vdW-DF-cx0), and revised VV10-and the strongly constrained and appropriately normed (SCAN) meta-generalized gradient approximation (GGA). Overall, we find that revPBE+D3 and RPBE+D3 show the best balance of performance for both the lattice parameters and the CO2 binding enthalpy of m-2-m-Zn2(dobpdc). revPBE+D3 and RPBE+D3 predict the m-2-m-Zn2(dobpdc) lattice parameters to within 1.4% of experiment and predict CO2 binding enthalpies of -68 kJ/mol, which compare reasonably well with the experiment (-57 kJ/mol). Although PBE (-57.7 kJ/mol), vdW-DF1 (-49.6 kJ/mol), and vdW-DF2 (-44.3 kJ/mol) are also found to predict the CO2 binding enthalpy with good accuracy, they overestimate lattice parameters and bond lengths. The other functionals considered predict the lattice parameters with the same accuracy as revPBE+D3 and RPBE+D3, but they overbind CO2 by around 26-50 kJ/mol. We find that the superior performance of revPBE+D3 and RPBE+D3 is sustained for the formation enthalpy and the lattice parameters of ammonium carbamate, a primary product of the cooperative CO2 insertion in diamine-functionalized M2(dobpdc) MOFs. Moreover, we find that their performance is derived from their larger repulsive exchange contributions to the CO2 binding enthalpy than the other functionals at the relevant range of the reduced density gradient value for the energetics of CO2 adsorption in the m-2-m-Zn2(dobpdc) MOF. A broader examination of the performance of RPBE+D3 for the structural parameters and CO2 binding enthalpies of 13 diamine-functionalized Mg2(dobpdc) MOFs further demonstrates that RPBE+D3 successfully reproduces experimental CO2 binding enthalpies and reveals a logarithmic relationship between the step pressure and the CO2 binding enthalpy of the diamine-functionalized Mg2(dobpdc) MOFs, consistent with experiments where available. The results of our benchmarking study can help guide the further development of versatile vdW-corrected DFT methods with predictive accuracy.