First-Principles Calculations on Σ3 Grain Boundary Transition Metal Impurities in Multicrystalline Silicon
슈퍼관리자
2021-05-21
First-Principles Calculations on Σ3 Grain Boundary Transition Metal Impurities in Multicrystalline Silicon
-
Authors :
A. Suvitha, N. S. Venkataramanan, R. Sahara, H. Mizuseki, and Y. Kawazoe
-
Journal :
Jpn. J. Appl. Phys.
-
Vol :
49
-
Page :
04DP02
-
Year :
2010
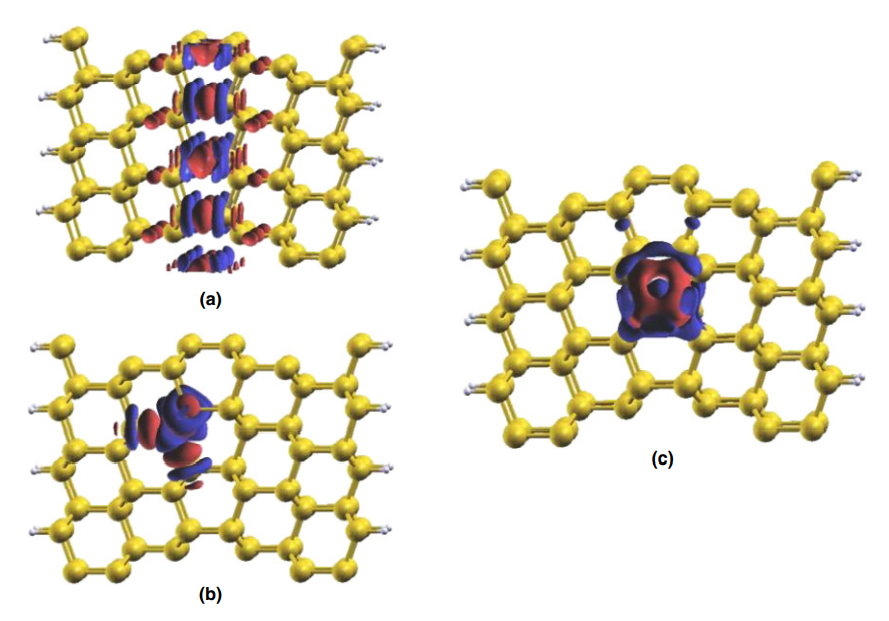
Abstract
We have carried out a density functional theory study on the Σ3 (111) silicon grain boundary, and calculated the impurity effect of Ni, Fe, Cu, and Cr atoms doped near the grain boundary at both interstitial and substitutional sites. The segregation energy for the impurities follows the order of Fe greater than Cu, Ni, and Cr at the substitutional site and Cr greater than Cu, Fe, and Ni, at the interstitial site. The calculated values were positive, indicating that segregation is not favored in the Σ3 (111) grain boundaries. When the metal impurity is placed at the substitutional site, a new state in the fundamental gap was observed in the density of states, the band gap is reduced, which may have an effect on the solar cell performance. The calculated magnetic moments for the transition-metal-doped grain boundary show that they were quenched for Ni, Fe, and Cu point defects.